

Software
SUPERFAMILY
There is a very low traffic mailing list for notification of updates/changes.
Domain-centric Gene Ontology
Gene Ontology terms have been mapped to SUPERFAMILY domains, supra-domains and architectures. The method has been assessed for its ability to predict GO terms on sequences in the CAFA competition. This dcGO also contains Phenotype Ontology terms and Anatomy ontology terms mapped from 8 different ontologies.
Database of disordered protein predictions
 D2P2 A community resource for pre-computed disorder predictions over a large library of known amino sequence. Goals of the database include making statistical comparisons of the various prediction methods freely available to the prediction community. As well as facilitating biological investigation of the disordered protein space.
D2P2 A community resource for pre-computed disorder predictions over a large library of known amino sequence. Goals of the database include making statistical comparisons of the various prediction methods freely available to the prediction community. As well as facilitating biological investigation of the disordered protein space.
Coiled Coil database
 Spiricoil is a website for Coiled Coil prediction in sequences. It also includes the option to view detected coiled coils mapped on known structures and even generate 3D models and view them. Spiricoil also includes the assignment of Coiled Coils to all completely sequenced genomes.
Spiricoil is a website for Coiled Coil prediction in sequences. It also includes the option to view detected coiled coils mapped on known structures and even generate 3D models and view them. Spiricoil also includes the assignment of Coiled Coils to all completely sequenced genomes.
SNP/SNV functional and phenotypic analysis
 FATHMM The Functional Analysis Through Hidden Markov Models (FATHMM) software and server is capable of predicting the functional and phenotypic consequences of protein missense variants using hidden Markov models (HMMs) representing the alignment of homologous sequences and conserved protein domains.
FATHMM The Functional Analysis Through Hidden Markov Models (FATHMM) software and server is capable of predicting the functional and phenotypic consequences of protein missense variants using hidden Markov models (HMMs) representing the alignment of homologous sequences and conserved protein domains.
Vector Graphics tree plotting
One of the main purposes of TreeVector is to move away from treating phylogenetic trees as end end point and final graphic, and to instead embed them in dynamic processes using web standard technologies, so that quick reference of a particular pattern or trait is possible, dynamic and up to date.
HMM model conversion
This script converts between the SAM and HMMER hidden Markov model formats. It was written as part of the project for comparing the performance of various profile comparison methods.Madera, M. and Gough, J. (2002). A comparison of hidden Markov model procedures for remote homology detection. Nucl. Acids Res., 30(19), 4321-4328.
HMM profile-profile comparison
 This is solely the work of Martin Madera in the Gough group. He wrote PRC the profile comparer. It is a stand-alone program for aligning and scoring two profile hidden Markov models.
This is solely the work of Martin Madera in the Gough group. He wrote PRC the profile comparer. It is a stand-alone program for aligning and scoring two profile hidden Markov models.
HMMER3 hmmscan sequence level threading
To make HMMER3 (actually only the hmmscan program) run faster on multiple processors/cores, use this hmmscan.pl wrapper. It requires the HMMER3 program 'hmmscan' of course. What it does is to run hmmscan on different sequences in different threads. USE AT YOUR OWN RISK. Please report ny problems you find with it. It is very easy to use, you just call it exactly the same way you call hmmscan, but specify the number of threads as an additional argument. We could have got it to auto-detect the available CPUs but this would cause a dependency on a library, so this feature was omitted.
Phylogenetic tree plotting
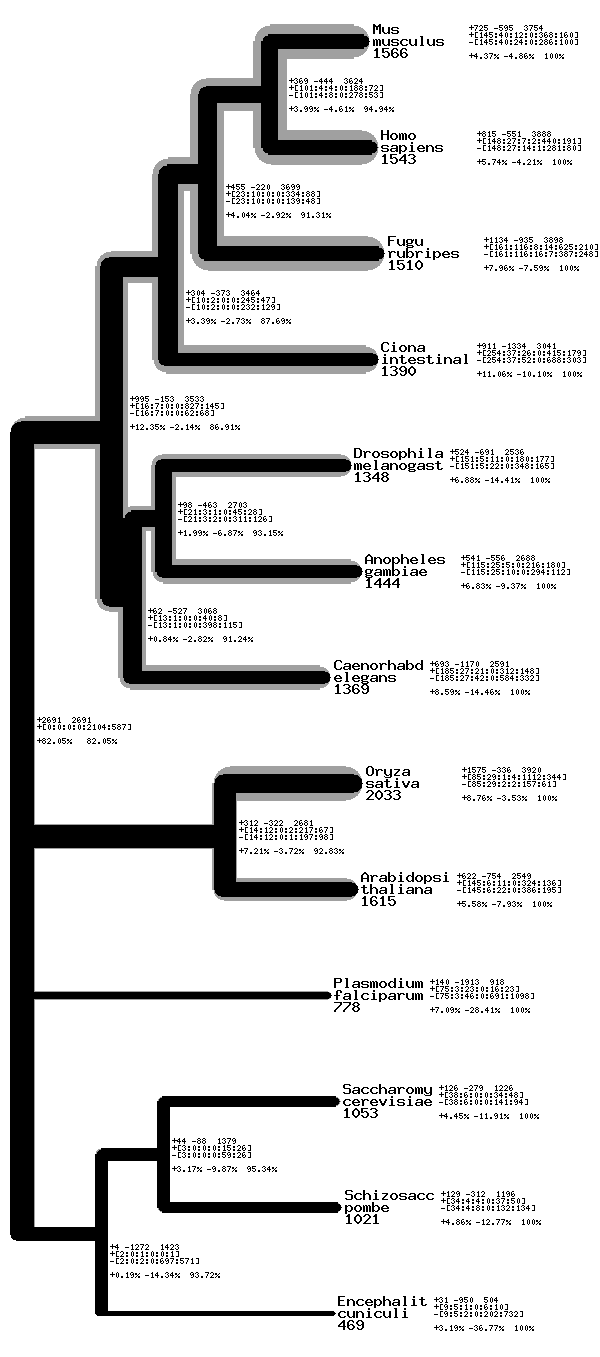
The module has a routine called ReadTree which reads the tree into two hash arrays. The first describes the tree by having a key for each node, where the value is the node above it in the tree. The second is the x-axis position of each node which gives the branch lengths.
There is also a drawing routine. In its simplest form the program merely draws a simple tree, with the user specifying the width and size of the image and the branches. It is possible however using hash arrays to specify two widths for each individual branch (one is grey, see example) and also multi-line labels for each node. There is in addition to this an extra label for each final member.